
Concept explainers
Videos
Adermatoglyphia (described previously in Problem 18 in Chapter 3) is an extremely rare condition where people are born without fingerprints; only four families on earth are known to have this condition. The condition is inherited in an autosomal dominant fashion and is due to point mutations in a gene on chromosome 4 called SMARCAD1.
The following figure shows that different point mutations—all near the 5′ end of the same intron of SMARCAD1—were found in each of the four families. All four mutations prevent the expression of a skin-specific transcript that uniquely contains exon 1, the first exon of this transcript; no other SMARCAD1 mRNAs contain this exon. In the figure, the final three bases in the RNA-like strand of exon 1 are shaded, while the first six bases of intron 1 are unshaded.
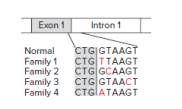
| a. | No ATG sequence normally exists in exon 1 upstream of the sequence shown. Which part of the skin-specific mRNA corresponds to exon 1? |
| b. | What aspect of gene expression is likely to be affected most directly by these mutations? |
| c. | Are these mutations more likely to cause loss of function or gain of function? |
Want to see the full answer?
Check out a sample textbook solution
Chapter 8 Solutions
Genetics: From Genes to Genomes


- Friedreich ataxia (FRDA) is an autosomal recessive, neurodegenerative disease that causes a lack of voluntary coordination of muscle movements. Affected individuals are homozygous for an unusually large number (expansion) of repeats of a trinucleotide sequence (GAA) in the first intron of the X25 gene. Unaffected individuals typically have between 7 and 38 repeats of the trinucleotide (GAAGAAGAAGAA…). FRDA patients have anywhere from 66 to over 1,700 repeats. To understand how the GAA trinucleotide expansion leads to FRDA, researchers looked at X25 gene expression by extracting RNA from affected and unaffected patients and doing a northern blot analysis (see the figure below): In panel “a,” the researchers used a probe to detect X25 mRNA. In panel “b,” the researchers used a probe on a duplicate of the original blot to detect human GAPDH mRNA (GAPDH is an enzyme involved in glycolysis). The sample labeled “YR” is mRNA from yeast cells that was used as a control. Explain…arrow_forwardThe D1S80 locus is located on human chromosome 1 and is characterized by a repeating 16 base pair (bp) sequence. Alleles for this locus vary depending on the number of repeats present, thus affecting the size of the locus. The D1S80 locus also contains two conserved sequences, a 32bp sequence at one end and a 113bp sequence at the other end. If the DNA of an individual is targeted for D1S80 amplification, and one of the resulting amplicons is approximately 785bp in size, how many repeats would be present in this D1S80 allele? The amplicon of interest is indicated by a red arrow in the diagram below.arrow_forwardThe genetic alteration responsible for sickle-cell anemia in humans involves: a transition mutation from A to G, substituting glutamic acid for valine in a-globin a transversion mutation from T to A, substituting valine for glutamic acid in b-globin a transition mutation from T to C, substituting valine for glutamic acid in b-globin a transversion mutation from G to C, substituting glutamic acid for valine in a-globin a frameshift mutation of one ATC codon, removing glutamic acid from b-globinarrow_forward
- Cx is a member of the family of connexin genes that encode the proteins of gap junction intercellular channels. Cx proteins in one cell form hemi-channels in the plasma membrane. Hemi-channels in adjacent cells dock to form complete intercellular channels through which ions and small molecules diffuse from cell to cell. Distinct Cx mutations were identified in three different families, F1, F2 and F3, affected by the same disease. To study their functional properties, normal (wild type, wt) and mutant (m) Cx proteins were expressed in cultured cells. Translation of the proteins was checked (Fig. 3). A extracellular EC 1 SM TM 1 membrane 2 3 4 F10 intracellular N F2 EC 2 F3 oricand c B 42 kDa C 42 kDa 35 kDa Control wt m-F1 m-F2 PM C PM C PM C PM C Western blot anti-Cx Control wt PM C m-F1 PM C PM C = Metabolically labelled m-F2 PM C m-F3 PM C m-F3 PM C WSEY Fig. 3 mont (A). Membrane topology of Cx protein indicating positions of mutations. Cx is an integral membrane protein with 4…arrow_forwardThere are five substitution mutations in the dark-colored mutant Mc1r gene. Compare the DNA sequence of the light-colored wild-type Mc1r gene with the DNA sequence of the dark-colored mutant Mc1r gene. Indicate the locations of the five mutations by changing the font color to YELLOW for the five single DNA nucleotides that are mutated in the mutant Mc1r gene table. Using the information in the introduction, determine whether each of these mutations is a silent, missense, or nonsense mutation. Using the mutant Mc1r gene data, fill in the columns (including DNA, mRNA, and amino acid) in gene table 2 that contain a silent mutation with BLUE. Likewise, fill in the columns that contain a missense mutation with RED. Shade any columns that contain nonsense mutations with GREEN. Then Of the five mutations you identified in the mutant Mc1r gene, how many are: substitutions insertions deletions (Enter a number on each line.) 2. Of the five mutations…arrow_forwardWhat is the underlying genetic defect that causes xeroderma pigmentosum?How can the symptoms of this disease be explained by the genetic defect?arrow_forward
- A gene contains 30% thymine. What is the percentage of pyrimidines present in this segment? Explain.arrow_forwardYou are interested in studying resistance to heavy metals and have selected the yeast Saccharomyces cerevisea to conduct your studies. You have recovered a deletion mutant that does not tolerate high concentrations of zinc (grows poorly in zinc containing media ) and have designated the mutant pgz-1 (for poor growth in zinc ). (a) What is the advantage to the type of mutant used in this work? What class of mutagen was likely use to generate pgz-1? ( b) Do you expect the PGZ gene to be expressed in your mutant? Explain.arrow_forwardHuntington’s disease is a hereditary central nervous system disorder characterized by tandem repeats of the sequence 5'-CAG-3' in the gene that encodes a protein called huntingtin. The disease is progressive from generation to generation, meaning that in later generations the number of CAG repeats increases and the age of onset of symptoms decreases. Refer to Figure 21.4 and describe the sort of evidence supporting the generational increase in the number of CAG repeats.arrow_forward
- STR sites are the basis of the FBI Laboratory's Combined DNA Index System (CODIS). One STR site that is used in CODIS is TPOX, named for its location within an intron of the thyroid peroxidase gene located on human chromosome 2. Different versions of the TPOX site are known, due to varying number of repeats of the short sequence "AATG". For TPOX, the number of repeats found on different chromosomes varies from 6 to 13. Hence the "alleles" of this site are called "6", "7", "8", "9", "io", "11", "12" and "13". Any given individual can be homozygous for any one of these eight different alleles or heterozygous for two different alleles of this set. The frequency of any given genotype, however, depends upon the individual frequency of each of these eight alleles in the population. For example, allele "6" occurs at a frequency of less than 5%, allele "11" at a frequency of 20% and allele 8" at a frequency of about 46%. See below for the double stranded DNA sequence of the *11" allele and a…arrow_forward1. a)What would happen if the aminoacyl tRNA synthetase responsible for charging alanine tRNAs also charged methionine tRNAs with alanine? b)What would happen if an individual was homozygous for mutant alleles of the gene encoding the aminoacyl tRNA synthetase responsible for charging leucine tRNAs?arrow_forwardThe following is a list of mutational changes. For each of the specific mutations described, indicate which of the following terms could apply, either as a description of the mutation or as a possible cause. More than one term from the right column can apply to each statement in the left column. 1. an A-T base pair in the wild-type gene is changed to a G-C pair 2. an A-T base pair is changed to a T-A pair a. transition b. base substitution c. transversion 3. the sequence AAGCTTATCG is changed to d. inversion AAGCTATCG c. translocation f. deletion 4. the sequence AAGCTTATCG is changed to AAGCTTTATCG g. insertion 5. the sequence AACGTTATCG is changed to AATGTTATCG h. decamination 6. the sequence AACGTCACACACACATCG is i. X-ray irradiation changed to AACGTCACATCG j. intercalator 7. the gene map in a given chromosome arm is changed from bog-rad-fox1-fox2-try-duf (where foxl and fox2 are highly homologous, recently diverged genes) to bog-rad-fox1-fox3- fox2-try-duf (where fox3 is a new gene…arrow_forward
 Human Anatomy & Physiology (11th Edition)BiologyISBN:9780134580999Author:Elaine N. Marieb, Katja N. HoehnPublisher:PEARSON
Human Anatomy & Physiology (11th Edition)BiologyISBN:9780134580999Author:Elaine N. Marieb, Katja N. HoehnPublisher:PEARSON Biology 2eBiologyISBN:9781947172517Author:Matthew Douglas, Jung Choi, Mary Ann ClarkPublisher:OpenStax
Biology 2eBiologyISBN:9781947172517Author:Matthew Douglas, Jung Choi, Mary Ann ClarkPublisher:OpenStax Anatomy & PhysiologyBiologyISBN:9781259398629Author:McKinley, Michael P., O'loughlin, Valerie Dean, Bidle, Theresa StouterPublisher:Mcgraw Hill Education,
Anatomy & PhysiologyBiologyISBN:9781259398629Author:McKinley, Michael P., O'loughlin, Valerie Dean, Bidle, Theresa StouterPublisher:Mcgraw Hill Education, Molecular Biology of the Cell (Sixth Edition)BiologyISBN:9780815344322Author:Bruce Alberts, Alexander D. Johnson, Julian Lewis, David Morgan, Martin Raff, Keith Roberts, Peter WalterPublisher:W. W. Norton & Company
Molecular Biology of the Cell (Sixth Edition)BiologyISBN:9780815344322Author:Bruce Alberts, Alexander D. Johnson, Julian Lewis, David Morgan, Martin Raff, Keith Roberts, Peter WalterPublisher:W. W. Norton & Company Laboratory Manual For Human Anatomy & PhysiologyBiologyISBN:9781260159363Author:Martin, Terry R., Prentice-craver, CynthiaPublisher:McGraw-Hill Publishing Co.
Laboratory Manual For Human Anatomy & PhysiologyBiologyISBN:9781260159363Author:Martin, Terry R., Prentice-craver, CynthiaPublisher:McGraw-Hill Publishing Co. Inquiry Into Life (16th Edition)BiologyISBN:9781260231700Author:Sylvia S. Mader, Michael WindelspechtPublisher:McGraw Hill Education
Inquiry Into Life (16th Edition)BiologyISBN:9781260231700Author:Sylvia S. Mader, Michael WindelspechtPublisher:McGraw Hill Education