
Concept explainers
Videos
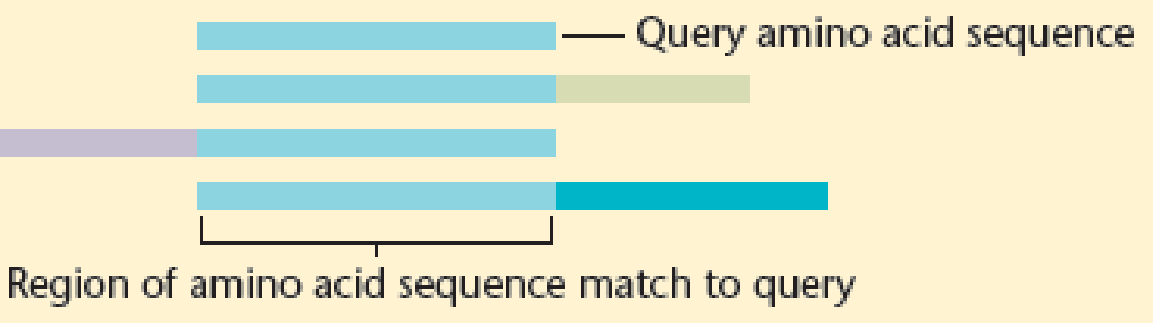
Annotation of a proteome attempts to relate each protein to a function in time and space. Traditionally, protein annotation depended on an amino acid sequence comparison between a query protein and a protein with known function. If the two proteins shared a considerable portion of their sequence, the query would be assumed to share the function of the annotated protein. Following is a representation of this method of protein annotation involving a query sequence and three different human proteins. Note that the query sequence aligns to common domains within the three other proteins. What argument might you present to suggest that the function of the query is not related to the function of the other three proteins?
Want to see the full answer?
Check out a sample textbook solution
Chapter 21 Solutions
Concepts of Genetics (12th Edition)


- Sequence: CCACCTGTACCCGGACACACCCTGGTGTCC What human disease has been connected to this gene? Calculate molecular weight (kiloDalton, kD) and calculated pI (the pH where the protein carries no net electrical charge) of the protein.arrow_forwardBelow is a sequence of 540 bases from a genome. What information would you use to find the beginnings and ends of open reading frames? How many open reading frames can you find in this sequence? Which open reading frame is likely to represent a protein- coding sequence, and why? Which are probably not functioning protein-coding sequences, and why? Note: for simplicitys sake, analyze only this one strand of the DNA double helix, reading from left to right, so you will only be analyzing three of the six reading frames shown in Figure 19.4.arrow_forwardSequence: CCACCTGTACCCGGACACACCCTGGTGTCC 1. Identify the gene from which the querysequence originates (Name of gene) 2. Provide the FULLprotein sequence encoded by the gene. 3. Are different splice variants known for this gene? 4. What human disease has been connected to this gene? 5. Calculate molecular weight (kiloDalton, kD) and calculated pI (the pH where the protein carries no net electrical charge) of the protein.arrow_forward
- I are studying pancreatic islet cells and have isolated, cloned, and sequenced a novel protein that you postulate has 4 transmembrane segments. Explain why the sequence would lead to this hypothesis (what procedure would I have applied).arrow_forwardConsider the following coding 71 nucleotide DNA template sequence (It does not contain a translational start): 5’- GTTTCCCCTATGCTTCATCACGAGGGCACTGACATGTGTAAACGAAATTCCAACCTGAGCGGCGT GTTGAG-3’ By in vitro translating the mRNA, you determined that the translated peptide is 15 amino acids long. What is the expected peptide sequence in single letter abbreviations?arrow_forwardKnowing that the genetic code is almost universal, a scientist uses molecular biological methods to insert the human - globin gene (shown in the figure below (Links to an external site.)) into bacterial cells, hoping the cells will express it and synthesize functional - globin protein. Instead, the protein produced is nonfunctional and is found to contain many fewer amino acids than does -globin made by a eukaryotic cell. Explain why and give thoughts as to how to overcome this.arrow_forward
- The following RNA sequence represents a small messenger which can be translated in a prokaryotic cell: 5'-ACGAAUGCACAGUAAAACUGGCUAGCGUAGGCUGA-3 Assume that the messenger RNA is translated in the cell, using the correct machinery and signals required for accurate protein synthesis. Using this RNA sequence and the Genetic Code Dictionary (see your textbook for the dictionary), solve the following problems A. Write the sequence of a protein that would be translated from this mRNA, using the appropriate stop and start signals, and indicating the correct termini of the protein product. B. Suppose that the underlined A in the sequence is changed to a U. Write the expected protein product of this mRNA.arrow_forwardMass spectrometry is a powerful tool in proteomics. What are the four key features of a mass spectrometer? Describe briefly how MALDI and two-dimensional polyacrylamide gel electrophoresis could be used to identify a protein expressed in cancer cells but not in normal healthy cells.arrow_forwardProteins called molecular chaperones assist in the process of protein folding. One class of chaperones found in organisms from bacteria to mammals is heat shock protein 90 (Hsp90). All Hsp90 chaperones contain a 10 amino acid signature sequence that readily allows identification of these proteins in sequence databases. Two representations of the Hsp90 signature sequence are shown here. Y-x-[NQHD]-[KHR]-[DE]-[IVA]-F-[LM]-R-[ED]. 4 YSNKE/FLRE 3. 7. 1 1 2 3 4 5 6 7 8 9 10 C Bitsarrow_forward
- In addition to in the information given in the pictures, researchers provide further descriptions for the figure stating: Players were also able to restructure b-sheets to improve hydrophobic burial and hydrogen bond quality. Automated methods have difficulty performing major protein restructuring operations to change b-sheet hydrogen-bond patterns, especially once the solution has settled in a local low-energy basin. Players were able to carry out these restructuring operations in such scenarios as strand swapping (Fig. 3) and register shifting. In one strand-swap puzzle, Foldit players were able to get within 1.1 A° of the native structure, with the top-scoring Foldit prediction being 1.4 A° away. A superposition between the starting Foldit puzzle, the top-scoring Foldit solution, and model 1 of the native NMR structure 2kpo (Protein Data Bank) are shown in Fig. 3b. Rosetta's rebuild and refine protocol, however, was unable to get within 2 A ° of the native structure (Fig. 3a, yellow…arrow_forwardDescribe the typical principles used to identify topogenic sequences within proteins and how these principles can be used to develop computer algorithms. How does the identification of topogenic sequences lead to prediction of the membrane arrangement of a multipass protein?arrow_forwardWhich of the following statements are true? Electrostatic interactions are the dominant forces in protein molecular recognition. When two proteins form a complex there is an unfavorable loss of rotational-translational entropy. Protein-protein interfaces are most often dry. The exclusion of water results in an unfavorable loss in rotational-translational entropy. The free energy change associated with the formation of an enzyme-substrate complex almost always results in an unfavorable reduction in conformational entropy of the proteins. Burial of an uncompensated positive charge inside proteins is usually unfavorable. So-called van der Waals’ interactions are essentially electrostatic in origin. Steric complementarity of the two partners forming a complex is essential to achieve optimal free energy of binding. Structural models of proteins obtained from low temperature crystallography are excellent descriptions of all biochemically relevant aspects of their function.arrow_forward
 Biology: The Dynamic Science (MindTap Course List)BiologyISBN:9781305389892Author:Peter J. Russell, Paul E. Hertz, Beverly McMillanPublisher:Cengage Learning
Biology: The Dynamic Science (MindTap Course List)BiologyISBN:9781305389892Author:Peter J. Russell, Paul E. Hertz, Beverly McMillanPublisher:Cengage Learning